MD Format — Metabolite is the Mass Dynamics format for uploading metabolite-level quantitative data.
This format is intended for metabolomics datasets where each metabolite or feature has a quantitative intensity value for each sample. It is typically used for discovery metabolomics and relative quantification workflows
File structure
MD Format — Metabolite uses a long-format table.
This means each row represents one metabolite, or feature, measured in one sample.
A wide matrix, where metabolites are rows and samples are columns, must be converted into long format before upload.
Required files
A metabolite upload requires:
| File | Required | Purpose |
|---|---|---|
| Metabolite intensity file | Yes | Contains metabolite or feature intensities for each sample |
experiment_design.csv |
Yes | Maps samples to the experimental design |
sample_metadata.csv |
Yes | Contains sample-level annotations |
The SampleName values in the metabolite intensity file must match the sample_name values in experiment_design.csv and sample_metadata.csv exactly.
Sample names are case-sensitive.
Required columns in the metabolite intensity file
Your metabolite intensity file must contain the following columns:
| Column | Type | Description |
MetaboliteId |
Text | Stable identifier for the metabolite or feature |
MetaboliteIntensity |
Numeric | Non-logged quantitative intensity value |
SampleName |
Text | Sample identifier |
Imputed |
Integer | 0 for measured values, 1 for missing or imputed values |
Column details
MetaboliteId
MetaboliteId is the identifier used to group rows belonging to the same metabolite or feature.
You can use another stable identifier such as:
| Identifier type | Example |
| InChIKey | Structure-based identifier |
| ChEBI | Metabolite identifier |
| HMDB | Metabolomics database identifier |
| KEGG Compound | Compound and pathway identifier |
| PubChem CID | Cross-reference identifier |
| Internal feature ID | Stable identifier from your workflow |
The identifier does not need to be the common metabolite name. In many cases, it is better to use a stable database ID or feature ID as MetaboliteId, then add a separate MetaboliteName column for readability.
The most important rule is consistency. If you want to compare or map metabolites across datasets, use the same identifier system across those datasets wherever possible.
MetaboliteIntensity
MetaboliteIntensity is the quantitative value for a metabolite or feature in a sample.
Upload non-logged intensity values.
You may upload raw, normalised, or analysis-ready intensity values, depending on your workflow. Do not upload log-transformed values. Normalisation and imputation can also be performed in Mass Dynamics after upload.
SampleName
SampleName identifies the sample associated with each intensity value.
Each SampleName must match the corresponding sample_name in:
| Companion file | Matching column |
experiment_design.csv |
sample_name |
sample_metadata.csv |
sample_name |
The values must match exactly, including capitalisation, spacing, and punctuation.
Imputed
Imputed tells Mass Dynamics whether the value is a measured value or a missing/imputed value.
| Value | Meaning |
0 |
Measured value |
1 |
Missing or imputed value |
The Imputed column must only contain 0 or 1.
Missing values
Missing measurements should be included in the file as:
| Field | Value |
MetaboliteIntensity |
0.0 |
Imputed |
1 |
Do not leave missing measurements blank.
Do not omit rows for missing measurements.
Do not use Imputed = 0 for a zero value unless you are certain the zero represents a real measured value.
In most metabolomics workflows, a value of 0.0 should be treated as missing and marked with Imputed = 1.
Full matrix requirement
The metabolite intensity file must contain a complete matrix.
This means every MetaboliteId must have exactly one row for every SampleName.
For example, if you have:
| Number of metabolites or features | Number of samples | Required rows |
| 3 | 4 | 12 |
| 100 | 20 | 2,000 |
| 1,000 | 50 | 50,000 |
A missing measurement is still included as a row. It should be represented as MetaboliteIntensity = 0.0 and Imputed = 1.
Visual example of the long format
If your wide matrix looks like this:
| MetaboliteId | MetaboliteName | Sample_A | Sample_B | Sample_C |
| HMDB0000122 | Glucose | 15320.5 | 0.0 | 14220.7 |
| HMDB0000094 | Citrate | 8421.3 | 9105.9 | 8770.2 |
It should be converted into long format:
| MetaboliteId | MetaboliteName | SampleName | MetaboliteIntensity | Imputed |
| HMDB0000122 | Glucose | Sample_A | 15320.5 | 0 |
| HMDB0000122 | Glucose | Sample_B | 0.0 | 1 |
| HMDB0000122 | Glucose | Sample_C | 14220.7 | 0 |
| HMDB0000094 | Citrate | Sample_A | 8421.3 | 0 |
| HMDB0000094 | Citrate | Sample_B | 9105.9 | 0 |
| HMDB0000094 | Citrate | Sample_C | 8770.2 | 0 |
In this example, the Glucose measurement for Sample_B is missing, so it is uploaded as 0.0 with Imputed = 1.
Extra metabolite metadata columns
You can include extra metabolite annotation columns in the intensity file.
Examples include:
| Column | Purpose |
MetaboliteName |
Human-readable metabolite or feature name |
Description |
Short description |
HMDB |
HMDB identifier |
KEGG |
KEGG Compound identifier |
ChEBI |
ChEBI identifier |
PubChemCID |
PubChem Compound ID |
mz |
Mass-to-charge value |
RetentionTime |
Retention time |
Formula |
Molecular formula |
SMILES |
Structure string |
Pathway |
Pathway annotation |
AnnotationStatus |
Identification or annotation confidence |
Extra columns are imported as metabolite-level metadata.
The value in each metadata column must stay the same for every row with the same MetaboliteId.
For example, if MetaboliteId = HMDB0000122 has MetaboliteName = Glucose, then every row for HMDB0000122 must also have MetaboliteName = Glucose.
Only MetaboliteIntensity and Imputed should vary between samples for the same MetaboliteId.
Example experiment_design.csv
sample_name,condition
Sample_A,Control
Sample_B,Control
Sample_C,Treatment
Example sample_metadata.csv
sample_name,condition,batch,timepoint
Sample_A,Control,1,Baseline
Sample_B,Control,1,Baseline
Sample_C,Treatment,2,Week_4
You can include additional sample-level annotations in sample_metadata.csv, such as batch, dose, timepoint, sex, tissue, treatment, or disease state.
Anything that varies by sample belongs in sample_metadata.csv, not in the metabolite intensity file.
Converting a wide matrix into MD Format — Metabolite
If your data is currently a wide matrix, you need to convert it into long format before upload.
Example Python conversion:
________________________________________________________________________________________________
import pandas as pd
wide = pd.read_csv("my_metabolomics_wide.tsv", sep="\t")
id_col = "MetaboliteId"
meta_cols = ["MetaboliteName", "Description"]
sample_cols = [
col for col in wide.columns
if col not in [id_col, *meta_cols]
]
long_df = wide.melt(
id_vars=[id_col, *meta_cols],
value_vars=sample_cols,
var_name="SampleName",
value_name="MetaboliteIntensity",
)
long_df["MetaboliteIntensity"] = long_df["MetaboliteIntensity"].fillna(0.0)
long_df["Imputed"] = (long_df["MetaboliteIntensity"] == 0).astype(int)
long_df.to_csv(
"md_format_metabolite.tsv",
sep="\t",
index=False
)
________________________________________________________________________________________________
This produces one row per metabolite or feature per sample.
Upload checklist
Before uploading, check that:
- The file contains
MetaboliteId,MetaboliteIntensity,SampleName, andImputed MetaboliteIntensityvalues are non-loggedImputedonly contains0or1- Missing values are represented as
0.0withImputed = 1 - Every metabolite or feature appears once for every sample
- Extra metabolite metadata columns are consistent within each
MetaboliteId SampleNamematchessample_nameinexperiment_design.csvandsample_metadata.csvexperiment_design.csvis includedsample_metadata.csvis included
Known limitations
Pathway enrichment and entity mapping depend on the identifiers provided in your upload.
Mass Dynamics links entities using the IDs and metadata supplied with the data. For best results across multiple uploads, use stable and consistent identifiers across datasets.
Steps to upload your metabolomics data using the MD Format
- Go to the Dataset Upload page
- Choose "Choose Files" or "Choose Folder" depending on how your files are stored
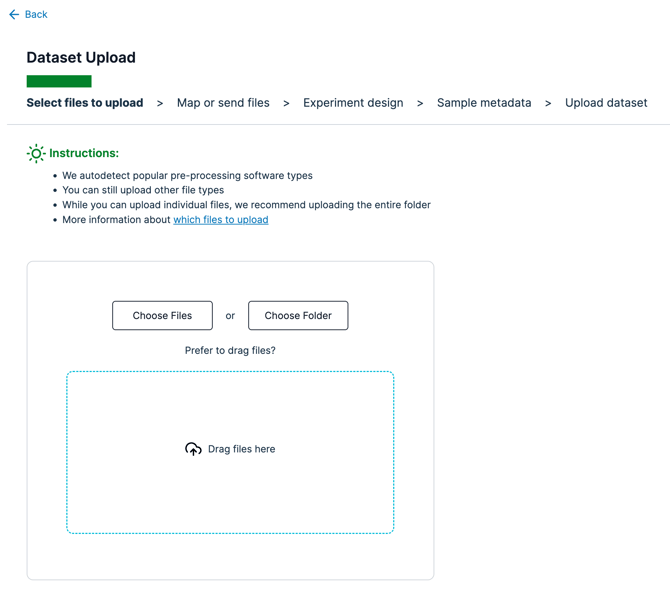
-
There is 1 file that is needed. Do not be concerned with the file name, as we will detect the data in the files based on the column names. Please ensure that your file contains your data in the following structure and that the files are uploaded with the "tsv" or “csv” file extensions.
- After choosing the relevant files to upload for your file system, click "Next"

- Mass Dynamics should now detect that it is the MD Format that you are using, and will detect the number of files found.

- You can now upload your own populated sample metadata csv file or simply click next and upload sample metadata later on.

- Go ahead and enter your Experiment Name and Experiment Description
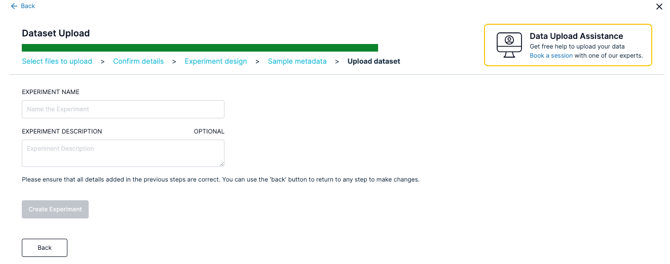
- Click "Create Experiment" and wait until your files upload
- Get started with analyzing your data!